This web page was produced as an assignment for Genetics 564, an undergraduate course at UW-Madison
Phylogeny
Phylogeny refers to the evolutionary history of organisms through time [1]. Through phylogeny and homology (see homologs tab), we can use similarities to order and organize what we are studying (whether it be organisms, genes, or proteins) in the form of a phylogenetic tree or phylogram. A phylogram allow us to visualize the evolutionary relationship between those members of our study. As the fossil record is incomplete, and constructing evolutionary history from living organisms is quite difficult, biotechnology computer programs have been developed to aid in reconstructing the evolutionary history of life [2]. There are three main types of reconstruction [2]:
Different online programs use different methods and algorithms to produce phylograms. No algorithm is perfect, and thus different programs result in different trees.
For this project, the FASTA sequences of the PER2 gene's mRNA and it associated homologs were entered into The European Bioinformatics Institute's program, Clustal Omega. Clustal Omega is a sequence-based method that infers trees from multiple sequence alignments [2]. To do this, it uses an algorithm to align sequences based on similarity and compares them to seeded guide trees. Clustal Omega can run large alignments and still maintain high accuracy [3].
- Distance methods: Characters (morphological structures, cell characteristics, biochemical pathways, genes, etc) are converted to distances. These distances are analyzed by an algorithm that uses neighbor joining or minimum evolution to construct a tree.
- Maximum parsimony: this method produces a tree that requires the least amount of character changes to explain the observed data.
- Likelihood methods: This method uses probability and maximum likelihood to produce a tree with the highest probability of fitting the data.
Different online programs use different methods and algorithms to produce phylograms. No algorithm is perfect, and thus different programs result in different trees.
For this project, the FASTA sequences of the PER2 gene's mRNA and it associated homologs were entered into The European Bioinformatics Institute's program, Clustal Omega. Clustal Omega is a sequence-based method that infers trees from multiple sequence alignments [2]. To do this, it uses an algorithm to align sequences based on similarity and compares them to seeded guide trees. Clustal Omega can run large alignments and still maintain high accuracy [3].
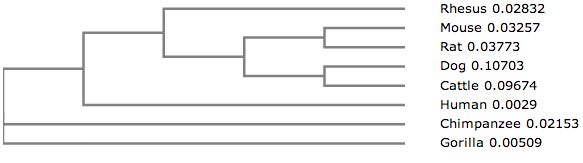
Phylogram from Clustal Omega
Analysis
From this phylogram, we can see that the chimpanzee and gorilla genes are closely related. The human gene is related to the chimpanzee and gorilla gene, but it shares a clade with the remaining organisms, indicating a common ancestor. The rat and mouse genes are most closely related, as well as the dog and cattle genes. As a whole, these four genes are most closely related to the rhesus monkey. These five genes share a clade, suggesting a common ancestor.
References
[1] Tree of Life Web Project. What is Phylogeny?
[2] Delsuc, F., Brinkmann, H., and Philippe, H. (2005). Phylogenomics and the reconstruction of the tree of life. Nature Review Genetics, 6, 361-375. doi:10.1038/nrg1603
[3] Sievers, F., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular System Biology, 7(539). doi:10.1038/msb.2011.75
[1] Tree of Life Web Project. What is Phylogeny?
[2] Delsuc, F., Brinkmann, H., and Philippe, H. (2005). Phylogenomics and the reconstruction of the tree of life. Nature Review Genetics, 6, 361-375. doi:10.1038/nrg1603
[3] Sievers, F., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular System Biology, 7(539). doi:10.1038/msb.2011.75
